• Polarity
• Electrical Charge
• Molecular Size
We are only going to be concerned with separation based on polarity since it is the most commonly used for chromatographic separation.
Separations Based on Polarity:
Organic molecules are sorted into classes according to the principal functional group(s) present in the molecule. Using a separation mode based on polarity , the relative degree of retention of different kinds of molecules is mainly determined by the nature and location of these functional groups.Classes of molecules can be arranged into a range of chromatographic polarity from highly polar to highly non-polar as shown below.

Polarity- based chromatography follows the rules which govern the partition of organic compounds between oil and water and which determines the partition coeffecient of a compound. In other words, water, being a small molecule with a high dipole moment, is a polar compound. On the other hand, benzene which is an aromatic hydrocarbon, is a non-polar compound. Molecules with similar chromatographic polarity tend to be attracted to each other and those with dissimilar polarity exhibit much weaker attraction and may even repel one another. This is the main frame for chromatographic separation modes based on polarity. Another way to think of this is : oil (non-polar) and water (polar) don’t mix. Chromatographic separations based on polarity depends upon the stronger attraction between likes and the weaker attraction between opposites. Remember, “Like attracts like” in polarity-based chromatography (the opposite of magnitism).
Based on the discussion above, in order to design a chromatographic separation system we create competition for the various components of the mixture between a mobile phase and a stationary phase with different polarities (thus forcing the different commponents to partition between the two pahses as they go through the column). Then, compounds in the sample that are similar in polarity to the stationary phase (column packing material) will be delayed because they are more strongly attracted to the particles. Compounds whose polarity is similar to that of the mobile phase will be preferentially attracted to it and move faster. Thus, the difference in the force of attraction of the different components of the mixture to the stationary and mobile phase causes separation between the analytes by changing the speed by which each analyte moves through the column. The larger the difference (between the components of the mixtrue) in the speed of movement the higher the selectivity of the chromatographic system.
Polarity of the mobile phase ranges between non polar molecules presented by hexane to the most polar molecule (water). Between the two solvents lies a range of molecules with varying degree of polarity as follows: water being most polar followed by the less polar methanol then acetonitrile, tetrahydrofuran then the least polar Hexane. We can always create mixtures with polarity that lies in between two solvents for example a mixture of water and methanol makes a solven mixture which is more polar then methanol and less polar than water. How much less polar than water will depend on the percentage of methanol in the mixture.When the analyte mixture is introduced on the column they become attracted to the particles of the stationary phase. A mobile phase which more effectively attracts the analyte will compete for it and displaces it from the stationary phase causing the analyte to move faster through the column (weakly retained).
For stationary phases which make the packing material of the column. Silica is the most polar, hydrophilic (water-loving) surface containing acidic Silanol ( silicon containing analog of alcohol) functional group. The activity or polarity of the silica surface may be modified selectively by chemically bonding to it less polar functional groups [bonded phase]. For examples the binding of cyanopropylsilyl- [CN] will give a less polar stationary phase then the less polar n-octylsilyl- [C8] then n-octadecylsilyl- [C18, ODS] mieties . The latter is a hydrophobic [water-hating], very non-polar packing.
After considering the polarity of both phases, then, a chromatographer must choose a mobile phase which will allow the analytes of interest to be retained by the stationary phase, to a certain extent, but not so strongly that they cannot be eluted. Among solvents of similar strength, the chromatographer considers which phase combination may best exploits of the little differences in polarity and solubility of the components of the mixture to maximize the selectivity ( separation between the eluted peaks) of the chromatographic system. But, as you probably can imagine from the discussion so far, creating a separation based upon polarity involves knowledge of the sample, and experience with various kinds of analytes and retention modes. Thus the chromatographer will choose the best combination of a mobile phase and stationary phase with sufficiently different polarities. Then, as the mixture of analytes moves through the column, the rule like attracts like will determine which analytes slow down and which move at a faster speed.

Normal-Phase HPLC :
A mixture of compounds was analized using a column packed with seilica (polar stationary phase) as shown above with a much less polar [non-polar] mobile phase. This mode of chromatography is known as normal phase.
The mixture is composed of three comounds. Compound A(yellow) which is very polar, Compound B (red) and is less polar and compound C (blue) is the least polar. SInce the stationary phase (silica) is polar thus it retains the polar yellow component most strongly. The relatively non-polar blue compound moves fastest down the column since it is the least polar so is the most attracted or retained by the mobile phase, a non-polar solvent, and elutes (comes out of the column) quickly.
Reversed-Phase HPLC:
The term reversed-phase describes the chromatography mode that is just the opposite of normal phase, namely the use of a polar mobile phase and a non-polar (hydrophobic) stationary phase. the figure above shows the same mixture of drugs separated before and which is composed of compound A (yellow) which is the most polar, compound B (red) of intermediate polarityand compound C (blue) which is the least polar in the mixture.
Now the most strongly retained compound is the more non-polar compound C (blue) , since it is the most attracted to the non-polar stationary phase. The polar yellow compound A, being weakly retained on the non polar stationary phase and is the nost attracted by the polar mobile phase, thus moves the fastest through the column packing, and elutes earliest (comes out of the column first) .
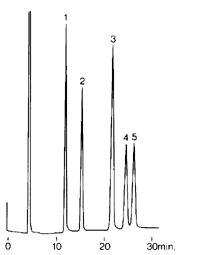
Reversed-phase chromatography is now concidered the most popular pahse of separation due to its higher reproducibility and ease of applicarion to a wide range of compounds. Now as each band (separated compund) is eleminated from the column it passes through the detector'e cell and gives a responce proportional to its concentration. The recorder will record a peak at a time corresponding to the time at which the particular band comes out (eluted) and the integrator will calculate the area or the hight of the peak which, in turn, is relative to the concentration of the band. Thus giving the known form of a chromatogram which is a plot of retention time vs. peak hight or area. In the case of reversed phase elemination of the yellow (1), red (2) and blue (3) compounds will give three well resolved peakson the chromatogram to the right.
Now as each band (separated compund) is eleminated from the column it passes through the detector'e cell and gives a responce proportional to its concentration. The recorder will record a peak at a time corresponding to the time at which the particular band comes out (eluted) and the integrator will calculate the area or the hight of the peak which, in turn, is relative to the concentration of the band. Thus giving the known form of a chromatogram which is a plot of retention time vs. peak hight or area. In the case of reversed phase elemination of the yellow (1), red (2) and blue (3) compounds will give three well resolved peakson the chromatogram to the right.
Given all the above facts and knowing the nature of the sample mixture being used the chromatographer is always facing the challenge of producing a chromatographic separation method which best suits his purposes. The chromatographer is always aiming at a chromatographic system which will produce an ideal chromatogram that will help him reproducibly analize his mixture.
References:
1-Laboratory outline and notebook for Pharamceutical Chemistry; Fourth year (802).
2-Instrumental methods of chemical analysis, Glen W. Ewing (1998).
3-Contemporary Instrumental analysis, Kennetha A. Rubinson and Sudith F. Rubinson.
4-Waters "the science of what is possible".
5-HPLC and CE principles and practice, Andrea Wetson.